Zespół Drave'a: przyczyny, objawy, rozpoznanie i leczenie
Zespół Drave'a jest oporną (nieleczoną) formą padaczki związaną z napadami wywołanymi gorączką. Charakterystyczny jest dziedziczony autosomalnie dominujący typ dziedziczenia.
Jest to ciężka napadowa padaczka miokloniczna (TMEM). Ta patologia jest również nazywana encefalopatią u noworodków. Występuje z częstością 1: 40000 noworodków, chłopcy chorują 2 razy częściej. Debiutuje w pierwszym roku życia i objawia się gorączką (wraz ze wzrostem temperatury) i gorączką (w normalnej temperaturze) uogólnionymi i miejscowymi napadami z obecnością krótkotrwałych tworów mięśniowych (mioklonia). Dzieci pozostają w tyle w rozwoju umysłowym, nie są podatne na leczenie przeciwpadaczkowe.
Klasyfikacja
Odnosi się do postaci padaczki, która ma objawy zarówno uogólnionej, jak i częściowej epilepsji.
Tło historyczne
Zespół został odkryty w 1978 roku przez epileptologa Charlotte Drave i nosi imię autora. Do tej pory tę formę padaczki przypisywano zespołowi Lennoxa i Gastauta.
Wielu autorów uważa nazwę TMEM za nieskuteczną, ponieważ napady miokloniczne zwykle łączą się kilka lat po debiucie, aw niektórych przypadkach mogą być nieobecne.
W 1999 r. Niemcy zaproponowały termin "ciężka padaczka idiopatyczna z objawami wielkiego mal w dzieciństwie".
W 2001 roku Japonia: hemi-grand mal dzieci.
W 2004 r. We Francji: padaczka polimorficzna wieku niemowlęcego.
Obecnie używany termin eponimicheskom - zespół Drave'a.
Etiologia
Czy patologia kanałów sodowych. Zwykle kanały sodowe utrzymują stałą ilość sodu w komórce i jej aktywność elektryczną. Zespół Drave'a rozwija się u osób z genetyczną wadą kanałów sodowych. Zjawisko to odkryto w 2001 r., Kiedy wyizolowano mutację genu SCN1A (podjednostka kanału sodowego). Od 2001 r. Kontynuowano badania nad mutacją genetyczną. Obecnie znanych jest ponad 100 rodzajów uszkodzeń genetycznych SCN1A.
Ad

Mutacje są różne i mogą zwiększać, zmniejszać lub nie zmieniać pobudliwości komórek nerwowych. Zespół Drave'a występuje tylko w przypadku zwiększonej pobudliwości neuronów i jest jedynym zespołem epileptycznym, którego diagnoza opiera się na analizie DNA.
W 20% przypadków mutacja genu nie jest przyczyną choroby, etiologia nie jest znana. Istnieją dowody na historię rodzinną drgawek i epilepsji w przebiegu gorączki.
Obraz kliniczny
Pierwszy objawy zespołu Drava może wystąpić w wieku 7 lat. Przebieg choroby jest wystawiony.
- Debiut estradowy. Powstaje w wieku 1 roku. Manifestowane w postaci drgawek gorączkowych. Występują z niską temperaturą i mają krótki charakter. Przekształcenie drgawek gorączkowych w różne formy padaczki wynosi od 3 do 5%.
- Etap "katastroficzny". W wieku 1-4 lat drgawki gorączkowe można zastąpić napadami padaczkowymi i padaczką.
- Stopień stabilizacji. Po 4-5 latach drgawki występują rzadziej? na czoło wysuwają się ogólne zaburzenia poznawcze (utrata pamięci, upośledzenie umysłowe) i deficyt neurologiczny (encefalopatia epileptyczna).
Stan neurologiczny
Przed wystąpieniem objawów klinicznych dzieci są zdrowe, a rozwój fizyczny i neuropsychiatryczny spełnia normy wiekowe. Po debiucie pojawia się zmniejszenie napięcia mięśniowego, odruchy ścięgien zwiększają się, pojawiają się objawy piramidalnej niewydolności. Po 1 roku dzieci mają ataksyjny (chwiejny) chód i dziwne ruchy.
Zaburzenia pozapiramidowe w postaci zwiększonego napięcia mięśniowego, drżenia i sztywności ruchów łączą się z wiekiem szkolnym. W dojrzewanie symptomatologia jest uzupełniona przez celowe drżenie (pojawia się podczas ruchu i jest nieobecny w spoczynku) i bez padaczki mioklonie (spontaniczne skurcze i rozluźnienie obszaru mięśniowego). Choroba jest komplikowana przez deformacje szkieletu (kifoscoliosis, flatfoot).
Ad
Postęp zaburzeń neurologicznych trwa aż do 8 lat, stan ustabilizuje się.
Rozwój poznawczy (wszystkie rodzaje aktywności umysłowej)
Na początku choroby rozwój psycho-mowy spowalnia, a później dochodzi do pełnej regresji. Równolegle występują zaburzenia zachowania (nadpobudliwość, autyzm). W wieku 5 lat rozwija się upośledzenie umysłowe. Poprzez zmniejszenie częstotliwości ataków możliwe jest poprawienie funkcji poznawczych.
Charakterystyka ataków
- Nietypowy absansy. Znaleziono u 40-95% dzieci. Atak rozwija się stopniowo na tle normalnych działań. Powstaje w wieku od 4 miesięcy do 6 lat. Charakteryzuje się on atonią (pominięcie ramion, bierne ukłony, nagły spadek) lub komponent miokloniczny (czynne rytmiczne kije, mioklonie powiek i mięśni obręczy barkowej). W towarzystwie wyraźnego zabarwienia wegetatywnego (odbarwienie skóry, zwiększona praca ślinianek i gruczołów potowych), zaburzenie chodu. Czas trwania od kilku godzin do kilku dni. Objawy są najbardziej widoczne po przebudzeniu.
- Masywne napady padaczkowe (mioklonie). Jest to główny objaw kliniczny i rozwija się rok lub dłużej po debiucie w wieku 1,5-4 lat. Stopniowo zwiększa się intensywność i częstotliwość ataków. Mioklonie najczęściej obserwuje się w mięśniach szyi i pleców. Widać to poprzez "kiwanie głową", potrząsanie ramionami z "wyrzucaniem" ramion, rytmiczne odchylanie głowy do tyłu, zginanie ciała. Dla ataków charakteryzujących się seriality, wzrost po przebudzeniu i zasypianiu, wysoka wrażliwość na światło.
- Bezgatunkowe mioklonie. Rozwija się po 4-5 latach. Jest sprowokowany przez ruchy. Obejmuje końce kończyn i ma niską amplitudę. Skurcze mięśni są wizualnie trudne do zauważenia, ale można dotykać. Nie ma zmian w EEG.
- Naprzemienne hemokonwulsje. Występują na jednej lub drugiej stronie ciała.
- Ataki ogniskowe. Znaleziono u 43-79% dzieci. Istnieją złożone silniki, automotory, paroksyzmy versus.

Rozpoznanie zespołu Drave'a
Trudny stopniowy rozwój obrazu klinicznego i debiut z drgawkami gorączkowymi. Od pojawienia się objawów do powstania diagnozy potrzeba 1-2 lat Diagnoza jest bez wątpienia w typowej klinice choroby. Można podejrzewać zespół Drave'a u każdego normalnie rozwijającego się dziecka z długotrwałymi drgawkami gorączkowymi, które rozpoczęły się w pierwszym roku życia i które rozwinęły się po napadzie padaczkowym dowolnego rodzaju i zaburzeniu rozwojowym.
Ad
Rozpoznanie opiera się na danych z badań klinicznych, analizie napadów padaczkowych, zmianach EEG i długotrwałej obserwacji pacjenta.
Kryteria diagnozy:
- Począwszy od 1 roku życia.
- Drgawki gorączkowe.
- Napady miokloniczne po 2 latach.
- Nietypowe, bezogonowe, ogniskowe, hemocywulsyjne i naprzemienne hemokonwersje.
- Wysoka częstość napadów, stan padaczkowy.
- Wyraźne naruszenia aktywności umysłowej.
- EEG. Istnieje stopniowy rozwój zmian EEG. W pierwszym roku życia wskaźniki odpowiadają normie, w drugim roku życia ujawniają spowolnienie głównej aktywności, połączenie regionalnej i rozproszonej aktywności padaczkowej, przejawy wrażliwości na światło. Obecnie monitorowanie snu wideo EEG staje się coraz bardziej diagnostyczne.
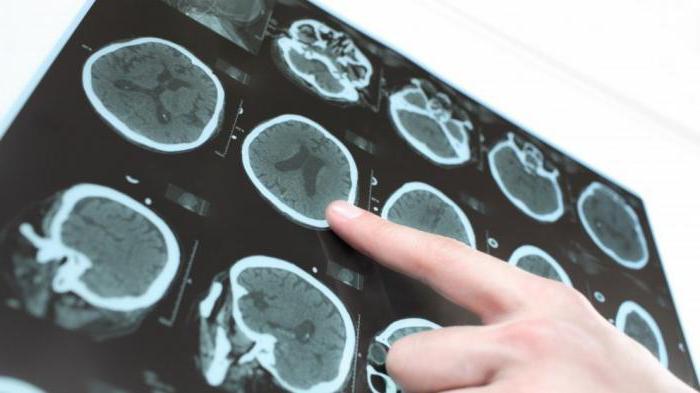
- Neuroobrazowanie z MRI: rozlane zaniki kory mózgowej, które jest bardziej wyraźne w płatach czołowych i skroniowych; zanik móżdżku; umiarkowany wzrost komory mózgu. Opisano pojedyncze przypadki w postaci zmian miejscowych: torbiele, stwardnienie skroniowe, zmniejszenie objętości płata skroniowego, dezhemelinację.
- Analiza DNA: wykrycie mutacji w genie SCN1A, której może brakować w zespole Drave'a.
- Autosomalny dominujący tryb dziedziczenia. Jeśli istnieje możliwość konsultacji z genetyką.
Leczenie zespołu Drave'a
TMEM jest jednym z najbardziej opornych na leczenie zespołów. Nie ma wysoce skutecznych leków przeciwdrgawkowych do kontrolowania napadów w zespole Drava. W młodym wieku barbiturany i benzodiazepiny mogą być skuteczne przez krótki czas; w starszym wieku połączenie walproinianu i topiramatu jest optymalne.
Leki przeciwpadaczkowe o pozytywnym działaniu:
- Lek z wyboru - "Valproat". Zwiększa zawartość GABA w ośrodkowym układzie nerwowym.
- Benzodiazepiny. Zmniejsz pobudliwość neuronów dzięki powinowactwu z hamującym neuroprzekaźnikiem GABA.
- Barbiturany. Hamuj transmisję impulsów nerwowych.
- "Ethosuximide". Hamuje niszczenie GABA, zmniejsza przewodnictwo impulsów nerwowych.
- Topiramat. Zwiększa aktywność GABA.
Leki przeciwpadaczkowe mogące zaostrzyć atak:
Ad
- "Lamotrygina".
- "Fenytoina".
- "Karbamazepina".
- "Wigabatryna".
Leczenie kortykosteroidami, immunoglobulinami, dietą ketogenną i stymulacją nerw błędny nie wykazało skuteczności.
Bardzo ważne jest monitorowanie temperatury ciała, szczególnie w okresie choroby zakaźnej. Wykazano, że takie dzieci obniżają temperaturę powyżej 37 stopni przyjmując leki przeciwgorączkowe. Monitorowanie temperatury powinno odbywać się rano i wieczorem. Konieczne jest wykluczenie przegrzania dziecka (gorące kąpiele, ekspozycja na słońce). Rodzice powinni wiedzieć o fotostymulacji (ekspozycja na światło). Niektóre dzieci mają autostymulację i czerpią z tego przyjemność. Powodują atak, zakrywając oczy i podnosząc gałki oczne w górę rytmicznym skurczem powiek. Ta grupa dzieci doświadcza obsesje otrzymując tak wątpliwą "przyjemność" i medyczną ulgę w atakach, nie pozostawiają one prób ponownego ataku.

Prognoza
Remisja jest niezwykle trudna do osiągnięcia. Celem leczenia jest zmniejszenie liczby napadów, zapobieganie epizodom seryjności i rozwój stanu padaczkowego. Konieczne jest poprawienie wskaźników funkcji poznawczych, dostosowanie dzieci w społeczeństwie. W wieku 12-14 lat ataki stają się rzadsze, a zaburzenia asteniczno-wegetatywne i zaburzenia psychiczne pojawiają się na czoło. Śmiertelność w wieku niemowlęcym wynosi 20%.