Zespół Klippel-Feila: objawy, zdjęcia, leczenie, rokowanie
W 1912 roku dwaj neurolodzy pochodzenia francuskiego, Andre Feil i Maurice Klippel, szczegółowo opisali wrodzoną deformację kręgosłupa szyjnego kręgosłupa, która pojawiła się u dzieci. Wszyscy pacjenci z tą chorobą byli dziećmi rodziców, którzy mają związek krwi.
Nie jest to zaskakujące, ponieważ w średniowieczu krewni mogli zostać legalnymi małżonkami. Nieco później ta patologia została nazwana na cześć naukowców. W rzeczywistości syndrom Klippel-Feil nie jest chorobą. Jest to wrodzona patologia, która może powodować rozwój innych chorób kręgosłupa. 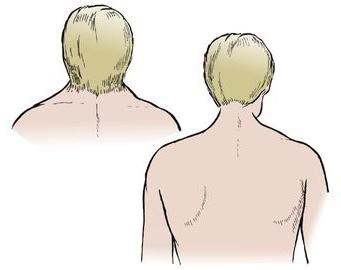
Cechy zespołu
Zespół ten jest wrodzoną patologią kręgosłupa szyjnego, która polega na zmniejszeniu liczby kręgów i synostozy. Występują dość częste objawy: obserwuje się je u jednego noworodka na 120 tysięcy. Najbardziej charakterystyczną cechą jest skrócenie szyi. Większość przypadków choroby towarzyszą inne patologie, które wpływają na układ mięśniowo-szkieletowy i narządy wewnętrzne. Rozpoznanie tej choroby leży w kompetencji tak wąskich specjalistów, jak neurolog, genetyk, ortopeda i kilku innych. Leczenie typu konserwatywnego może polegać na masażu, fizjoterapii i terapii ruchowej. Jeśli sprawa jest poważna, nie jest wykluczone zapotrzebowanie na leczenie chirurgiczne, polegające na szyjce macicy.
Należy rozróżnić defekty genetyczne od bardziej powszechnego zespołu kręczu szyi lub fałszywej szyi u nowo narodzonego dziecka, które wynika z urazu porodowego. Kiedy noworodek przemieszcza się wzdłuż kanału rodnego, kręg szyjny jest ściśnięty, co tworzy iluzję, że szyja jest krótka. Takie anomalie można korygować, zwykle znikają bez śladu w pierwszym roku życia.
Przyczyny syndromu Klippel-Feila
Zespół ten zalicza się do grupy chorób genetycznie zdeterminowanych. Zmiany chorobowe zaczynają się rozwijać w pierwszych tygodniach rozwoju płodu w łonie matki. Lekarze zwracają uwagę na kilka głównych powodów, w wyniku których objawia się zespół. Należą do nich zaburzenia segmentacji i rozwoju kręgosłupa, głównie na górnym poziomie szyjki macicy.
Co pomaga w określeniu obrazu klinicznego?
Obraz kliniczny choroby można określić:
- Awaria łuków i ciał.
- Zmniejszenie liczby kręgów.
- Synostozy kręgów szyjnych i piersiowych.

Grupa ryzyka obejmuje dzieci, które nie osiągnęły sukcesu, charakteryzujące się:
- Wady genetyczne w chromosomach. Dziecko jest zaburzone przez tworzenie zróżnicowania wzrostu, które jest niezbędne, aby szkielet rozwijał się w pełni. Naruszenie to wpływa na rozwój kręgów piersiowych i szyjnych.
- Dziedziczenie dziedziczone przez autosomalne. Oznacza to, że: jeśli jedno z rodziców cierpi na chorobę, wtedy chore dziecko urodzi się z prawdopodobieństwem 50-100 procent.
- Autosomalny recesywny typ dziedziczenia. W tym przypadku prawdopodobieństwo maleje i wynosi od zera do pięćdziesięciu procent.
Aby uniknąć dysfunkcjonalnego dziedziczenia, rodzice powinni zasięgnąć porady w zakresie genetyki przed poczęciem dziecka.
Objawy zespołu Klippel-Feila
Choroba ta charakteryzuje się klasyczną triadą objawów:
- Zmienia granice wzrostu włosów.
- Zaobserwowano zbyt skróconą szyję.
- Mobilność głowy jest ograniczona.

Najczęściej jest on łączony z innymi chorobami. Około trzydzieści procent pacjentów z zespołem Klippel-Feila cierpi na skoliozę, sztywne formy kręczu szyi i wysoki stan łopatek, zwany chorobą Sprengla. W niektórych przypadkach stwierdzone nieprawidłowości rąk, deformacje stóp, zęby. Obserwowane asymetria twarzy i dalekowzroczność. Około dwudziestu pięciu procent pacjentów cierpi na wrodzoną głuchotę.
Ten zespół jest nie tylko defektem kosmetycznym o jasnym nasileniu. Ponadto pod nim leżą poważne powikłania neurologiczne. Można je wyrazić w rozwoju oligofrenii, wodogłowia, epilepsji. Od wczesnego dzieciństwa pacjenci doświadczają osłabienia mięśni kończyn, synkinezy. W wieku nieco starszych, kliniczny obraz choroby jest uzupełniony pojawieniem się wtórnych zmian w kręgosłupie.
Jeśli dostępne objawy zespołu Klippel-Feil, musi zostać zbadany.
Badanie lekarskie
Zespół ten jest weryfikowany na podstawie obecnej triady objawów, badania instrumentalnego i badania fizykalnego. Szczególną rolę w diagnozie przypisuje się proces badania historii rodziny pacjenta. Zespół Klippel-Feila można zdiagnozować, a związane z nim anomalie można opisać za pomocą skoordynowanej pracy tak wąskich specjalistów jak kardiolodzy, neurologowie, ortopedzi, genetycy, pulmonolodzy.
Ocena charakteru zmian kręgosłupa pozwala na wykonanie radiografii. Takie badanie należy przeprowadzić w dwóch projekcjach, przy czym rzut boczny jest bardziej informacyjny. Z uwagi na fakt, że głowa jest nietypowa, cień czaszki może być połączony z obrazem kręgosłupa, który z kolei nie pozwala ujawnić szczegółów. Zaleca się również wykonywanie dodatkowych zdjęć, które wykazują maksymalne zgięcie i rozciągnięcie szyi. To one umożliwiają wykrycie niestabilności nieadherentnych kręgów. Co jeszcze zawiera diagnoza zespołu Klippel-Feila? 
Radiografia radiologiczno-klatki piersiowej kręgosłup pozwala wykryć:
- Obecność zdeformowanych kręgów.
- Akrecja ciał kręgów.
- Zmniejsz ich liczbę.
- Krzywizna kręgosłupa.
- Nieprawidłowa pozycja ostrzy.
Po potwierdzeniu rozpoznania lekarz przepisze dodatkową procedurę badania ultrasonograficznego narządów wewnętrznych. Pozwala to na identyfikację anomalii w ich obecności. Jeśli syndromowi towarzyszy patologia neurologiczna, konieczne może być przeprowadzenie ultrasonografii USG naczyń, MRI regionu szyjnego, EEG i angiografii. Konsultacja z genetyką jest obowiązkowa. Na podstawie uzyskanego wyniku lekarz jest w stanie określić, jaki rodzaj choroby jest dziedziczny i jakie jest ryzyko wystąpienia choroby w przyszłych pokoleniach.
Formy zespołu
Do chwili obecnej zespół Klippel-Feil (zdjęcia można zobaczyć w tym artykule) jest dość rzadką patologią. Objawy choroby rozpoznaje się u jednego dziecka na sto dwadzieścia tysięcy. Ta anomalia ma trzy formy: 
- Liczba segmentów okolicy szyjnej zmniejsza się, stopniowo rosną razem, w wyniku czego następuje wizualne skrócenie szyi. Ta forma choroby powoduje trudny ruch głowy.
- Powstała synostoza odcinka szyjnego kręgosłupa w wyniku fuzji z kością potyliczną. Dzięki tej formie pacjent traci zdolność odwracania głowy bez bólu. Kość potyliczna i kręgi szyjne stanowią jedną całość.
- Połączenie dwóch pierwszych rodzajów patologii jest trzecią formą.
Tylko specjalista, który uzyska wyniki niezbędnych badań, jest w stanie określić formę tego zespołu.
Zasady leczenia choroby
Niestety, współcześni lekarze nie mają odpowiednich metod do całkowitego wyeliminowania tego zespołu. Terapia obejmuje tylko zapobieganie występowaniu wtórnych deformacji. Najczęściej stosowane metody leczenia zachowawczego, które opierają się na masażu i terapii wysiłkowej. Terapia lekami może być przepisywana, gdy syndromowi towarzyszy manifestacja silnego bólu, aw takim przypadku, jeśli występuje ucisk korzeni nerwowych. Najczęściej stosowane są leki z grupy przeciwbólowej, a także leki o działaniu przeciwzapalnym. Jeśli kompresja zostanie przedłużona, wskazana jest interwencja chirurgiczna. W tym przypadku głównym celem operacji jest wyeliminowanie bólu i częściowa korekcja wad zewnętrznych.
Chirurgiczne leczenie zespołu Klippel-Feil
Głównym wskazaniem do zabiegu jest utrzymujący się ból, który powoduje kompresję korzeni nerwowych. Warto zauważyć, że wraz z wiekiem stan pacjentów znacząco się pogarsza. Dlatego operacja jest przeprowadzana niezwłocznie, natychmiast po rozpoznaniu zespołu. W celu zwiększenia ruchomości szyi pacjenta, stosując technikę, taką jak szyjkowanie, metodą Bonol. Taka operacja polega na usunięciu czterech górnych żeber i okostnej, co zmniejsza nacisk na narządy wewnętrzne. Przeprowadzić podobną operację w kilku etapach: po pierwsze chirurg usuwa żebra od pacjenta z jednej strony, po czym następuje dość długi okres rehabilitacji i odbudowy ciała. Następnie wykonywany jest drugi etap operacji, a żebra są usuwane z drugiej strony. 
Okres rehabilitacji jest bardzo długi i wymaga dużego nakładu pracy ze strony pacjenta, jego krewnych i lekarzy. Pacjent, który przeszedł taką interwencję chirurgiczną, musi spędzić kilka miesięcy w stanie stacjonarnym. Jednak nie przerywaj operacji. Współczesna medycyna może zapewnić tylko jedną skuteczną metodę leczenia, a to jest właśnie taka operacja. Eliminuje zewnętrzne wady. Jeśli jednak odrzuci się leczenie, możliwe jest wystąpienie powikłań zespołu Klippel-Feil.
Możliwe powikłania
Pierwszym z nich jest zaostrzenie chorób narządów wewnętrznych, a następnie rozwija silny zespół bólowy. Ból wynika z naruszenia korzeni nerwowych. To, przy okazji, może doprowadzić do całkowitego unieruchomienia.
Choroby narządów wewnętrznych zagrażają nieodwracalnym procesom patologicznym. Rezultatem może być wczesna śmierć pacjenta. 
Prognoza
Jakie są prognozy dla zespołu Klippel-Feila?
Jeśli operacja i okres rehabilitacji powiodą się, pacjent może liczyć na pełne życie. W wyniku operacji nastąpi wizualne wydłużenie szyi, co jest korzystne z estetycznego punktu widzenia.
Środki zapobiegawcze
Jak już zauważyliśmy, zespół ten jest genetycznie zdeterminowany, dlatego nie ma specyficznej profilaktyki tej choroby. Jeśli zdarzały się przypadki przejawów podobnej patologii w rodzinie, obowiązkiem jest odwiedzić genetyka na etapie planowania dziecka. Specjalista przeprowadzi niezbędne badania, oceni ryzyko wystąpienia zespołu Klippel-Feil u nienarodzonego dziecka. Konsekwencji można uniknąć.