Zespół Marfana: objawy, rozpoznanie, przyczyny, objawy i leczenie
Zespół Marfana przejawia się różnymi patologiami szkieletu, układu sercowo-naczyniowego, zaburzeniami narządu wzroku. Ta choroba jest dziedziczna, jest wrodzona, a tkanka łączna ciała cierpi na nią.
Co uderza najpierw
Zespół Marfana jest naruszeniem normalnego funkcjonowania tkanki łącznej. Ponieważ rozwija się niepoprawnie, wpływa na wiele narządów wewnętrznych. Pacjenci mogą odczuwać następujące nieprawidłowości w ciele:
- gigantyzm;
- tętniak aorty;
- dolichostenomelia i arachnodaktylia;
- krótkowzroczność;
- ektopia obiektywu;
- kifoskolioza;
- odkształcenie mostka;
- płaskie stopy;
- ektazja opony twardej;
- występ panewkowy.
Istnieją zatem wspólne cechy fizyczne u osób, u których zdiagnozowano to (zespół Marfana). Zdjęcia pacjentów wykazują podobieństwo zewnętrznych objawów choroby, a specjalista zrozumie wiele z wyglądu pacjenta. Tymczasem, ze słabym objawem tej choroby, zewnętrzne objawy mogą być prawie niewidoczne. 
Jak diagnozuje się?
Ta choroba w praktyce medycznej jest rzadkością. Według ekspertów tylko 10-20 tysięcy osób może mieć zespół Marfana. Diagnostyka opiera się na kilku metodach. Jest to historia rodzinna, a także badania funkcji ciała, badania oczu, prześwietlenia i prognozy genetycznej. Jednocześnie nie ma różnicy, jaką płcią jest dana osoba, do jakiej rasy należy i gdzie się ona znajduje. Choroba ta dotyka zarówno mieszkańców południa, jak i północy.
Jeśli lekarz zdiagnozuje zespół Marfana, leczenie może być zarówno zachowawcze, jak i chirurgiczne. Konieczne jest odwołanie się do interwencji chirurgicznej w poważnych przypadkach, gdy leczenie lekami i procedurami fizjologicznymi nie przynosi pożądanych rezultatów.
Chirurgia jest często poddawana układowi sercowo-naczyniowemu i narządom wzroku.
Przyczyny choroby
Dlaczego występuje zespół Marfana? Przyczyny tkwią w mutacjach genu FBN1. To on jest odpowiedzialny za syntezę fibryliny. To strukturalne białko nadaje tkance łącznej niezbędną kurczliwość i elastyczność. Z niedoborem fibryliny w ciele źle uformowane struktury włókniste. Tkanka łączna tracąc jednocześnie swoją siłę i elastyczność. Jest o wiele gorszy w stanie wytrzymać stres fizjologiczny. Od tego początku cierpią narządy wewnętrzne i szkielet osoby. 
Kto najbardziej ryzykuje
Jak już wspomniano, zespół Marfana, którego oznaki mogą pojawić się już w pierwszych latach życia danej osoby, jest dziedziczony. Jeśli cierpiał na tym ktoś z rodziny choroba genetyczna Szanse są wysokie, że dziecko może również odziedziczyć tę chorobę.
Ale zdarza się również, że płód nadal ma pierwotną mutację w łonie matki. W tym przypadku im starsza kobieta, tym większe ryzyko zachorowania na chore dziecko. Dotyczy to szczególnie kobiet, które zajdą w ciążę po 35 latach.
Zewnętrzne manifestacje
Najczęściej tę chorobę można zauważyć nawet przy pomocy zewnętrznych znaków. Co więcej, im starszy staje się pacjent, tym wyraźniejsze są zewnętrzne cechy charakterystyczne. U osób, które mają historię zespołu Marfana, zdjęcie, jak już wspomniano, jest bardzo podobne.
Ich szkielet ma pewne cechy - ciało jest stosunkowo krótkie, wysokie, kończyny długie i cienkie, nieproporcjonalne do szkieletu. Palce są również wydłużone, pajęczaki. 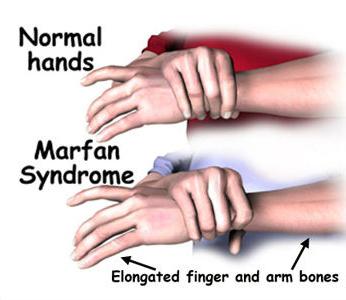
Zespół Marfana ma objawy takie jak:
- asteniczna budowa ciała;
- niedostatecznie rozwinięta tkanka podskórna;
- hipotonia mięśni;
- wąski i długi szkielet twarzy (dolichocephaly);
- wysokie łukowate niebo, jak również naruszenie ukąszenia (prognathia).
Przy narodzinach dziecka z zespołem Marfana długość ciała u chłopców wynosi ponad 53 cm, u dziewczynek - 52,5 cm, a ostateczna wysokość odpowiednio w okolicy 192 i 175 cm, są też wyższe.
Dotkliwość
Ta choroba ma różny stopień uszkodzenia. W związku z tym osoby z rozpoznanym zespołem Marfana mogą mieć różne objawy.
Lekarze przydzielają kilka różnych postaci tej choroby, w zależności od tego, na jakie systemy życia są narażone.
Pierwsza forma jest wymazana. Zmiany i nieprawidłowości w rozwoju są łagodne. Tylko jeden lub dwa systemy ciała cierpią, a nie bardzo wiele.
Druga postać zespołu Mafrana jest wyraźna. Istnieje kilka opcji tutaj:
- Zmiany są łagodne, ale obecne w trzech systemach ciała.
- W jednym systemie występują oczywiste odchylenia od normy.
- Zmiany są wyraźnie wyrażone w dwóch, trzech lub więcej systemach.
Lekarze zauważają również trzy ciężkości choroby: łagodną, umiarkowaną i ciężką. Przebieg choroby może być inny. Istnieje stabilny zespół Marfana, którego oznaki w ciągu lat obserwacji pozostają niezmienione. Istnieje progresywny typ choroby, gdy z czasem patologie rozwojowe rosną i się pogarszają.
Niektóre cechy choroby
Jak już zauważyliśmy, zespół Marfana charakteryzuje się:
- połączone uszkodzenia szkieletu, oczu, układu sercowo-naczyniowego i nerwowego;
- różnorodność objawów fizjologicznych;
- pierwsze oznaki choroby można rozpoznać zarówno po urodzeniu, jak i po latach;
- przewlekły postępujący przebieg.
W tej chorobie obserwuje się utrzymującą się dysfunkcję stawów. Eksperci nazywają ten stan nadmierną ruchliwością, gdy stawy są łatwo skręcone w różnych kierunkach i bardzo mobilne.
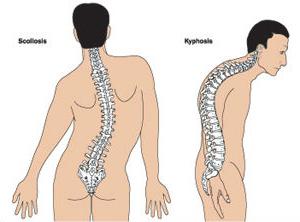
Wielu pacjentów cierpiących na zespół Marfana ma nieregularną budowę klatki piersiowej, przyjmuje postać w kształcie lejka lub kila.
Ortopedzi często zauważają deformację kręgosłupa w różnych jego przejawach:
- skolioza;
- kifoskolioza;
- kifoza;
- dyslokacje i podwichnięcia regionu szyjnego;
- kręgozmyk.
Pacjenci często cierpią na płaskostopie i wysunięcie panewki.
Powikłania serca
Patologia układu sercowo-naczyniowego często dominuje w obrazie klinicznym tej choroby. Przejawia się na różne sposoby. Pacjent może doświadczać uporczywych defektów w ścianach naczyń krwionośnych. Tracą elastyczność. Aorta, duże gałęzie tętnicy płucnej są szczególnie podatne na to. Pacjenci mogą doświadczać wad rozwojowych aparatu zastawkowego i ścian serca.
Jeśli chodzi o aortę, najczęściej obserwuje się progresywną ekspansję jej wstępującej części i pierścienia zastawki (poszerzenie, ektazja annuloaortalna). Pacjent często ma tętniaki, dotyczy to zastawki mitralnej. Istnieje również patologiczne wydłużenie struny i ich pęknięcie.
U dziecka, które wciąż pozostaje w macicy, ale na poziomie genów, zespół Marfana już został mu przekazany, często powstają wrodzone wady serca. Mogą to być:
- koarktacja aorty ;
- ubytek przegrody międzyprzedsionkowej (ASD);
- zwężenie płucne;
- wada przegrody międzykomorowej (VSD).
Mogą również wystąpić zaburzenia rytmu serca (tachykardia, migotanie przedsionków), rozwój infekcyjnego zapalenia wsierdzia.
W najbardziej niekorzystnej postaci noworodkowej zespołu Marfana, niewydolność serca rozwija się gwałtownie od samego urodzenia dziecka. Często te dzieci nie żyją do wieku jednego.
Upośledzona funkcja wzrokowa
Pacjenci z tą chorobą w większości przypadków cierpią na patologię narządu wzroku. Mogą to być:
- krótkowzroczność;
- spłaszczenie i zwiększenie rozmiaru rogówki;
- dyslokacja / podwichnięcie soczewki;
- hipoplazja tęczówki i mięśnia rzęskowego;
- zmiana kalibru naczyń siatkówki;
- strabismus.

W tym samym czasie, w wieku 4 lat, ektopia soczewki często rozwija się, rozwija się gwałtownie, funkcja wzrokowa pogarsza się.
Główne leczenie
Warto zauważyć, że z tą chorobą jeden lekarz nie może przepisać leczenia. Eksperci o innym profilu prowadzą obserwacje i wydają zalecenia: okulista, ortopeda, kardiolog, kardiochirurg, terapeuta, genetyk.
Przede wszystkim leczenie ma na celu zapobieganie progresji tej choroby. Ponieważ ostatecznie nie można pozbyć się tej choroby, ważne jest, aby ją powstrzymać, aby w przyszłości nie wpływać na narządy życiowe.

Jeśli wystąpi niewydolność zastawek serca, zostanie przedstawiona operacja. Z pilną potrzebą załóż sztuczną zastawkę mitralną.
Kobiety w ciąży, których zespół Marfana ma wyraźną patologię sercowo-naczyniową, kieruje się na wczesne cięcie cesarskie.
Jeśli widzenie jest osłabione, eksperci wybierają specjalne okulary i soczewki kontaktowe. W przypadku oczywistych patologii, gdy diagnozy takie jak zaćma, jaskra, przemieszczenie soczewki są wykonywane, zalecane jest leczenie chirurgiczne lub laserowe.
Ponadto operacja jest pokazana, jeśli w układzie kostnym występują wyraźne naruszenia. Lekarze utrzymują stabilizację kręgosłupa, torakochirurgię, alloplastyka stawu biodrowego.
Ilu pacjentów żyje z tą chorobą
W przypadku zespołu Marfana prawdopodobieństwo nagłej śmierci jest wysokie. Tylko specjaliści mogą przewidzieć, jak długo dana osoba będzie żyć z taką chorobą, biorąc pod uwagę stopień uszkodzenia ciała. Terminowe leczenie i korekcja kardiochirurgiczna mogą poprawić jakość życia i wydłużyć jego czas do 60-70 lat. Nie należy zapominać, że osoby z zespołem Marfana muszą przejść diagnozę i być stale monitorowane przez lekarza.
Lekarze ograniczają aktywność fizyczną do pacjentów z tą chorobą, są przeciwwskazani do dużych obciążeń, nurkowania, sportów kontaktowych.
Kobiety, które mają historię tej choroby i planują mieć dzieci, powinny zostać zbadane przez genetyka.