Zespół Pierre Robina: przyczyny, leczenie, konsekwencje. Chirurgia szczękowo-twarzowa u dzieci
Zespół Pierre'a Robina został nazwany na cześć francuskiego dentysty, który odkrył w 1923 r. Związek z manifestacją objawów tej choroby. Chociaż w XIX wieku niektóre z nich zostały opisane w literaturze medycznej. Choroba jest dość rzadka, ale statystyki różnią się nieznacznie w zależności od kraju. Tak więc w Europie (Dania) z powyższą patologią pojawia się 1 dziecko spośród 14 000 noworodków, które przeżyły poród, a w USA jedno dziecko z 20 000-30 000 urodzonych dzieci. Przejawem choroby jest defekt w okolicy szczękowo-twarzowej człowieka.
Przyczyny choroby
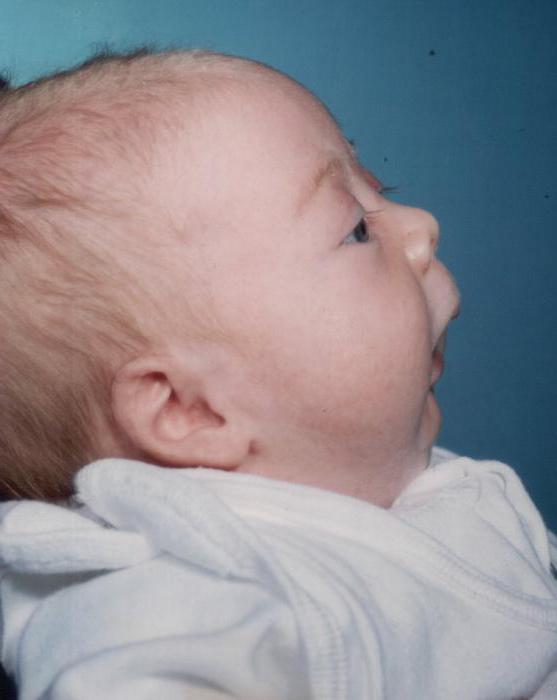
Zaburzenia rozwojowe dolna szczęka a podniebienie występuje podczas wzrostu płodu dziecka. Chociaż nie wszystkie czynniki zostały wyjaśnione, dzięki czemu pojawia się syndrom Pierre Robin. Patologia może wystąpić w różnych okolicznościach:
Ad
- W przypadku efektu infekcyjnego na wczesnym etapie ciąży matki.
- Podczas wstrząsów lub guzów w macicy, w czasie ciąży.
- W przypadku zaburzeń genetycznych może również pojawić się syndrom Pierre'a Robina (dziedziczenie wygląda tak: w około 5% przypadków drugie dziecko rodzi się z zespołem, jeżeli w rodzinie jest jedno chore dziecko).
- Czasami patologia objawia się w przypadku posiadania dwóch lub więcej dzieci.
Oznaki kliniczne

Głównym objawem choroby jest hipoplazja żuchwy - jej niedorozwój (takie dzieci mieć sfazowany, niedorozwinięty podbródek), w związku z czym jama ustna jest zmniejszona, a język jest cofany. Szczelina tworzy się na podniebiennych płytkach dziecka. Funkcja oddychania i połykania jest trudna. Bardzo często w zespole Pierre'a Robina pojawiają się inne patologie:
Ad
- Naruszenie struktury aparatu słuchowego, które pociąga za sobą utratę słuchu.
- Choroby ośrodkowego układu nerwowego (upośledzenie funkcji motorycznych, mikro- i wodogłowie, opóźniony rozwój mowy i psychiki, napady padaczkowe).
- Wrodzone anomalie funkcja wzrokowa - zaćma, krótkowzroczność.
- Choroba serca.
- Obecność dodatkowych palców na kończynach.
- Brak niektórych kończyn.
- Nieprawidłowo ukształtowany układ moczowy.
- Naruszenie formacji kręgosłupa i klatki piersiowej.
- Dzieci z zespołem Pierre Robina w 20% przypadków rodzą się z upośledzenie umysłowe.
U dziecka urodzonego z tą patologią oddychanie jest często chrapliwe i trudne, błony śluzowe i skóra stają się niebieskie. Jeśli nie pomożesz w czasie, może to być śmiertelne.
Typy chorób według stopnia trudności

Zespół Pierre'a Robina u noworodków dzieli się na kilka stopni, w zależności od zdolności do samodzielnego działania ciała:
- Łagodny typ choroby (dziecko ma normalny oddech, występują drobne problemy z połknięciem jedzenia, które matka może wyeliminować).
- Średni typ choroby (czynności oddechowe i połykanie są trudne, może wystąpić asfiksja). Wymaga leczenia w klinice szpitalnej.
- Ciężki rodzaj choroby (noworodek sam w sobie jest praktycznie niezdolny do oddychania, niemożliwe jest przyjmowanie pokarmu przez usta). W tym przypadku, aby przeżyć, dziecko wymaga pilnej interwencji chirurgicznej.
Leczenie zachowawcze
Na początku dzieci, które mają defekt w obszarze szczękowo-twarzowym, są karmione przez rurkę, stopniowo przechodząc na karmienie piersią. Leczenie zespołu ma na celu ułatwienie oddychania, uwolnienie dziecka od braku tlenu i wyeliminowanie przerwy w podniebieniu miękkim. Zdarzają się przypadki, gdy okazuje się, że poprawiają funkcję oddechową dziecka ze względu na wygodną pozycję ciała.
Ad
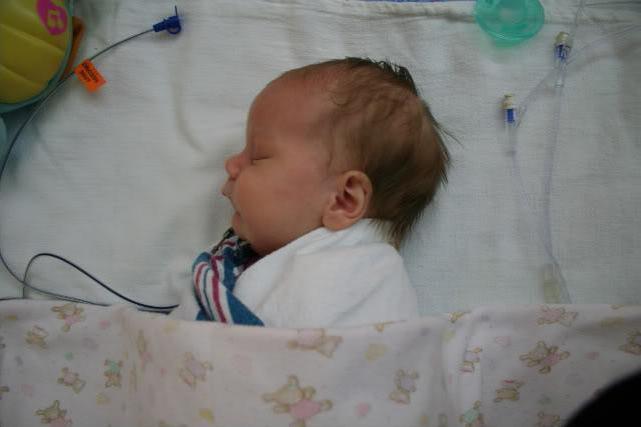
Leczenie może być zachowawcze i operacyjne, w zależności od złożoności choroby. W większości przypadków, przy łagodnym nasileniu, leczenie polega na umieszczeniu dziecka na brzuchu. Ta metoda przyczynia się do wzrostu żuchwy, która przesuwa język pacjenta dalej od tylnej ściany, co prowadzi do łatwiejszego oddychania. Kiedy dziecko porusza się lub płacze, jego oddech staje się lepszy, ale podczas snu znowu staje się trudne, dlatego konieczny jest stały nadzór nad takimi dziećmi.
Stosowane są również różne leki - przeciwdrgawkowe i uspokajające (Phenobarbital, Sibazon i inne). Po pierwszym miesiącu życia dziecka zaleca się przeprowadzenie neurosonografii (diagnostyki ultrasonograficznej mózgu) w celu wykrycia nieprawidłowości w jej strukturze.
Interwencja operacyjna
Jeśli zespół Pierre Robina ma umiarkowany stopień nasilenia, aby uniknąć interwencji chirurgicznej, dziecko wkłada plastikową płytkę na górną szczękę, blokując rozszczepione podniebienie i popychając język dziecka do przodu.

W zależności od konkretnego przypadku choroby, personel medyczny może wykonać tymczasową gloskopeksję (ruch języka z tylnej części jamy ustnej i jej przymocowanie do dolnej wargi dziecka). To operacja przetrzymywany przez 1-2 miesiące. Karmienie dziecka w tym czasie odbywa się przez rurkę gastrostomijną (otwór wykonany w żołądku do karmienia dziecka). Wyjście gastrostomii powstaje zwykle ze ścianek tkanki żołądka lub jelita cienkiego. Jest on wprowadzany, stale lub przez okres karmienia, gumową rurką, która służy jako sonda do wprowadzania pokarmu do żołądka.
Ad
Leczenie ciężkiej choroby
Współczesna chirurgia szczękowo-twarzowa leczy wszystkie stopnie choroby Pierra Robina.
W ciężkich przypadkach hipoplazja żuchwy dziecka, w ciężkich przypadkach zespołu, osteosynteza kompresyjna (chirurgiczna metoda wpływania na struktury kostne jest również stosowana w celu zapewnienia procesu osteogenezy - odbudowa i wydłużenie kości ludzkich) i tracheostomii (otwór oddechowy wykonany w tchawicy, gdzie rura), aby przywrócić samodzielność oddechową.
Do operacji osteosyntezy potrzebne są specjalne urządzenia do mocowania dolnej szczęki dziecka, jego ściskania i wydłużania. Istnieją urządzenia produkcji zagranicznej i tańszej produkcji krajowej. Urządzenie przymocowane jest do dolnej szczęki dziecka za pomocą dwóch kołków po prawej i lewej stronie. Między nimi kość jest przecinana i łączona przez kompresję przez 5 dni w celu utworzenia młodej kości. Szóstego dnia części szczęki powoli zaczynają się oddalać (około 1 mm na dzień). Metoda wydłużania młodej kości nazywana jest dystrakcją .
Ad
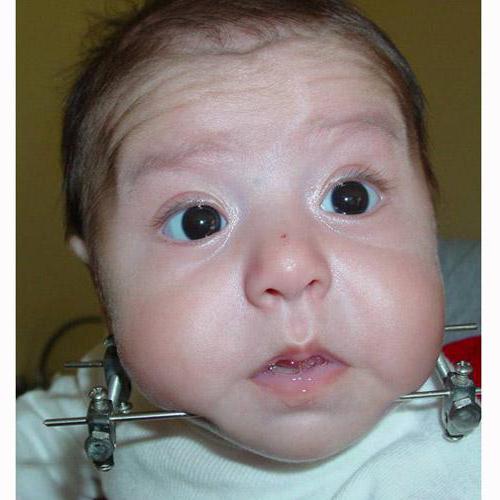
Podczas zabiegu stosuje się znieczulanie intubacyjne (tymczasowe odłączenie wrażliwości i świadomości osoby w mózgu, z porażeniem funkcji mięśniowych, a także wprowadzenie rurki oddechowej do wentylacji płuc).
Po 5-6 dniach od rozpoczęcia wydłużania żuchwy dziecka, najczęściej przywracana jest niezależna funkcja oddechowa dziecka. Rozproszenie uwagi następuje do momentu przywrócenia prawidłowej proporcji twarzy. Pod koniec tej procedury dziecko zostaje przeniesione do normalnego karmienia i wypisane do domu. Urządzenia usuwa się w znieczuleniu ogólnym, zaledwie 3 miesiące po ostatecznym przekształceniu wydłużonej części szczęki w twardą kość.
Usunięcie rozszczepu podniebienia
Operacja eliminacji rozszczepu podniebienia jest pożądana, zanim dziecko zacznie mówić (od 6 miesięcy do 1,5 roku). Czas trzymania zależy w dużej mierze od normalizacji oddychania dziecka. Przed uranoplastyką zaleca się noszenie urządzenia (obturatora) oddzielającego jamę nosową od jamy ustnej w celu normalnego oddychania i karmienia.
Chirurgia szczękowo-twarzowa jest zwykle przeprowadzana w kilku etapach:
- W pierwszym etapie następuje "rozwijanie" miękkiego podniebienia.
- W drugim etapie - plastikowe podniebienie twarde. Różni specjaliści z zakresu chirurgii szczękowo-twarzowej zalecali jednorazowe podejście do czasu etapów usuwania anomalii podniebienia górnego. Drugi etap uranoplastii można przeprowadzić od 1 roku do 10-11 lat.

Konsekwencje choroby
Jak już wspomniano, dzieci, które cierpiały na syndrom Pierre'a Robina, mogą nieco opóźnić się w rozwoju inteligencji i mowy, mogą mieć problemy z widzeniem i słyszeniem, a także istnieje możliwość chorób centralnego układu nerwowego. Ale leczenie rozpoczęte w całości i na czas może poprawić nie tylko wygląd, ale także życiową aktywność dziecka.